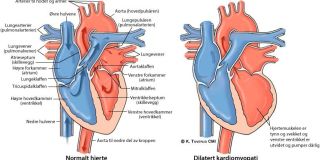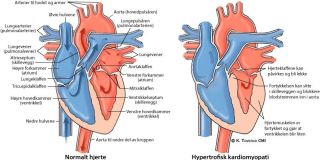

Ved hypertrofisk kardiomyopati (HCM) ses økt veggtykkelse av hjertemuskelen. I litt over halvparten av tilfellene med HCM kan man påvise en sykdomsgivende genfeil (mutasjon) som fører til forandringer i hjertemuskelcellens strukturer som har med hjertemuskelcellens sammentreknings-evne å gjøre. Dersom man tar en vevsprøve fra hjertemuskelen, ser man at hver enkelt muskelcelle er forstørret, og i enkelte områder er hjertemuskelcellene organisert i et uregelmessig mønster. Det er også økt mengde arrvev i hjertemuskelen, noe som sannsynligvis skyldes både redusert oksygentilførsel til hjertemuskelen og økt produksjon av arrvev.
Oftest er det hjerteskilleveggen mellom venstre og høyre hovedkammer som fortykkes mest. Dersom fortykkelsen sitter i venstre hjertekammers utløp, kan dette føre til at blodstrømmen hindres når blodet pumpes ut i kroppen. Dette kalles obstruktiv HCM. Hos pasienter med obstruktiv HCM kan man ofte høre en bilyd når man lytter til hjertet med stetoskop.
Dersom man kun er bærer av en mutasjon som kan gi HCM, uten å ha utviklet forstørret hjerte, har man som regel ingen symptomer. Pasienter med moderat eller betydelig grad av fortykket hjertemuskel, og da særlig obstruktiv HCM, blir i varierende grad slitne i forbindelse med anstrengelse, og kan også være plaget med brystsmerter. Pasienter med HCM har økt risiko for ulike rytmeforstyrrelser.
Det kan være aktuelt å starte behandling med betablokker for å redusere hjertefrekvensen og stabilisere hjerterytmen. Hos pasienter med obstruktiv HCM kan det i noen tilfeller også være nødvendig med kirurgisk septal myektomi, et hjertekirurgisk inngrep hvor man opererer bort en liten bit av det mest fortykkede området i hjerteskilleveggen. Alle pasienter med HCM vurderes med tanke på hvor stor risiko det er for alvorlige hjerterytmeforstyrrelser og om det er behov for implanterbar hjertestarter (ICD).
Vi oppfatter genbærere uten symptomer eller økt tykkelse av hjertemuskelen og uten arytmi-episoder som friske og disse får ingen restriksjoner når det gjelder fysisk trening. Dersom pasienten får en kardiomyopati-diagnose eller alvorlig arytmi, frarådes konkurranseidrett og høyintensitetstrening. I tillegg er helsekravene til førerkort gruppe 2 og 3 da ikke lenger oppfylt, og det er enkelte yrker som kan bli vanskelige å utøve.
Arytmogen kardiomyopati
Arytmogen kardiomyopati (AC) kjennetegnes av økt risiko for hjerterytmeforstyrrelser og svekkelse av hjertemuskulaturen, oftest i høyre hjertehalvdel.
AC skyldes en genfeil som gir endringer i desmosomene i hjertet. Desmosomer er proteiner som binder sammen hjertemuskelcellene med hverandre. Man kan forestille seg desmosomene som limet mellom cellene. Når desmosomene er svekket, vil mekanisk stress, som ved fysisk aktivitet, kunne føre til at limet mellom cellene ryker og hjertemuskelceller går til grunne. Disse hjertemuskelcellene blir erstattet av fett og bindevevsceller. Når deler av hjertet består av bindevev, er man mer utsatt for hjerterytmeforstyrrelser.
Det mest typiske er at sykdommen manifesterer seg i tidlig voksen alder. Dessverre kan alvorlig hjertearytmi og hjertestans være det første symptomet på AC, og sykdommen er den vanligste årsaken til plutselig død blant unge i Skandinavia. Svimmelhet og synkope, særlig i forbindelse med fysisk aktivitet, er symptomer man skal være oppmerksom på, da de kan være uttrykk for alvorlig hjerterytmeforstyrrelse. I senere stadier av sykdommen kan det utvikles hjertesvikt. Dette ses oftest hos eldre pasienter, men det finnes eksempler på unge AC-pasienter som har hatt behov for hjertetransplantasjon.
Medikamentell behandling, i form av betablokker, vurderes hos alle pasienter med AC for å minske risiko for arytmi. Det kan også bli aktuelt å operere inn en implanterbar hjertestarter. Denne registrerer hjerterytmen og kan avlevere livreddende sjokk ved en eventuell hjertestans.
Pasienter med AC frarådes å drive idrett på konkurransenivå. I perioder kan trening i det hele tatt frarådes.
Dersom et barn er bærer av en mutasjon som kan gi AC, men ikke har tegn til sykdommen, behandles det som hjertefriskt og får ingen restriksjoner angående fysisk aktivitet. Det anbefales imidlertid ikke at barnet satser på en idrettskarriere på sikt.